Key AI Techniques in small molecule screening

Introduction
Drug discovery is a lengthy, complex and costly process that comes with a high degree of uncertainty on whether or not a drug will succeed. During the drug discovery process, early stages rely on identifying drug candidates that bind to a known target (e.g. a protein). Because there are billions of possible drug candidates, it is very unlikely to find a good candidate by testing them one by one. As such, this process has been scaled up to test many compounds at once. This process is known as high throughput screening (HTS); while HTS happens in vitro, virtual HTS (vHTS) is based on in silico computations. HTS and vHTS are currently going through major innovations using Artificial Intelligence (AI). In fact, AI has prompted innovations in this field during the past decade and is predicted to continue doing so. In this article we explore key AI techniques applied in the early stages of drug discovery: deep learning, transfer learning, and active learning. Furthermore, we analyze the different challenges and benefits of each of these AI techniques.
Deep learning
What is deep learning?
Deep learning relies on the use of artificial neural networks which mimic the functioning of the brain. Neural networks actually learn by example as a child’s brain would. Classifying images, sounds, or texts into user-friendly categories is a typical task for deep learning models. Whether neurons are artificial or from a biological source, both operate as data processing units. Artificial neurons process bytes (digital information) in a network, while real neurons process electrical signals (biological information) in a brain. The shared etymology between deep learning and biology is therefore no coincidence.
A single layer of these artificial neurons is called a “shallow” network, while multiple layers that are stacked one on top of another is called a “deep” network. This network of layers is trained by feeding it data, and giving it feedback on the output that it generates from this data in order for the network to improve over time - i.e. a process of “learning”. These two concepts together explain the origin of the term “deep learning”. Deep learning networks tend to be able to better grasp complex, non-linear relationships between the input that they’re receiving and the output that is expected of them. This has proven in many use cases to enable more accurate representations of complex real-life processes. Undoubtedly, drug discovery also falls within this process category.
Applications of deep learning in drug discovery
Researchers in deep learning are currently implementing deep learning to drug discovery pipelines.
The 2013 Merck challenge appears as an early public indicator of the potential behind deep learning algorithms. In fact, the accuracy of Merck's In-house model was increased by 14% with a neural network, according to Gawehn et al., 2016 (Deep Learning in Drug Discovery). This challenge focused on drug property and activity prediction.
Yet, deep learning is also bringing innovations in other key components of the drug discovery process, such as :
- automated image analysis, using deep learning algorithms to extract data from digital images;
- automated in silico drug generation, using deep learning algorithms to design leads in drug discovery;
- protein structure prediction, using amino acid sequences to predict a protein’s 3D structure;
- chemical synthesis planning, to find reactions that will only affect one part of the molecule and leave the other parts unchanged.
Benefits of deep learning
Over the last decade, several groundbreaking milestones in computer vision and natural language processing have been achieved. The breakthroughs are mostly based on deep learning (DL) techniques. There is little doubt that such algorithms are going to break barriers in drug discovery too.
Deep neural networks take a shortcut by being able to quickly analyze small molecules instead of needing to go through lengthy and costly in silico analysis. While this is true for other machine learning algorithms, deep learning algorithms have a number of significant advantages.
For instance, traditional machine learning algorithms, such as random forests or support vector machines, fail at leveraging data that are not tabular, unless lossy preprocessing is implemented. On the other hand, richer data can be embedded into deep learning models. In fact, some researchers tackle the challenge of small molecule screening with deep graph neural networks. The work on Pushing the Boundaries of Molecular Representation for Drug Discovery with the Graph Attention Mechanism is one example. Since vHTS deals with molecules, modelization techniques should integrate 3D features in their design. Deep learning is a great tool to achieve this goal.
Deep learning models have also been challenging the need for feature engineering in some tasks. Feature engineering is the process of using domain knowledge to extract features from raw data. These features can be used to improve the performance of machine learning algorithms. In an impressive research, Zeiler & Fergus (2014) show that deep learning models can extract low-level features in images, such as edges and straight lines, and use them for accurate predictions. Associated together with the concept of transfer learning (see next section), automated feature extraction becomes a really powerful ally in deep learning.
Challenges in deep learning
Deep learning and Big Data are often referred to as being complementary to one another. In essence, deep learning works better with large volumes of data. They are indeed known for not plateauing in performance when the amount of data increases. On the contrary, their performance is expected to keep improving, according to the universal approximation theorem.
Luckily, in the space of drug discovery, large publicly available datasets for small molecule screenings are available. The DUD-E dataset and PubChem are good representatives. Even though such initiatives bear great promises, Schroedl (2019) in Current methods and challenges for deep learning in drug discovery notices that not only the volume but also the quality of molecule screening data is still far behind compared to other deep learning fields. The production of labeled data requires time-consuming and expensive in silico experiments. This ground reality may explain the data scarcity issue in small molecule screening. It is currently one of the most important challenges of deep learning in small molecule screening.
The lack of standardization is also a known challenge. Notably, the absence of unique identifiers for some chemical structures across data sources prevents smooth data integration. In lab experiments also follow different protocols. Hence, aggregating results of different trials might be very challenging. Some researchers work on curating publicly available datasets. In A new semi-automated workflow for chemical data retrieval and quality checking for modeling applications, the authors implement a curating workflow with guidelines towards reproducibility.
Overall, Big Data unleashes the full potential of deep learning models. Although, vHTS is lacking both volume and quality in its public resources nowadays. Standardization is also a core issue for datasets owned by corporations and curating mechanisms are often needed.
Transfer Learning
What is transfer learning?
Transfer learning is a mechanism used in deep learning where knowledge from a generic problem is transferred to a domain-specific problem. For instance, Convolutional Neural Networks trained on large-purpose image datasets, such as ImageNet, have been known for demonstrating great classification performance in more constrained settings (e.g. discriminating images of cats and dogs).
Applications of transfer learning in drug discovery
In Transfer Learning for Drug Discovery, the authors depict the process of deep transfer learning as follow:
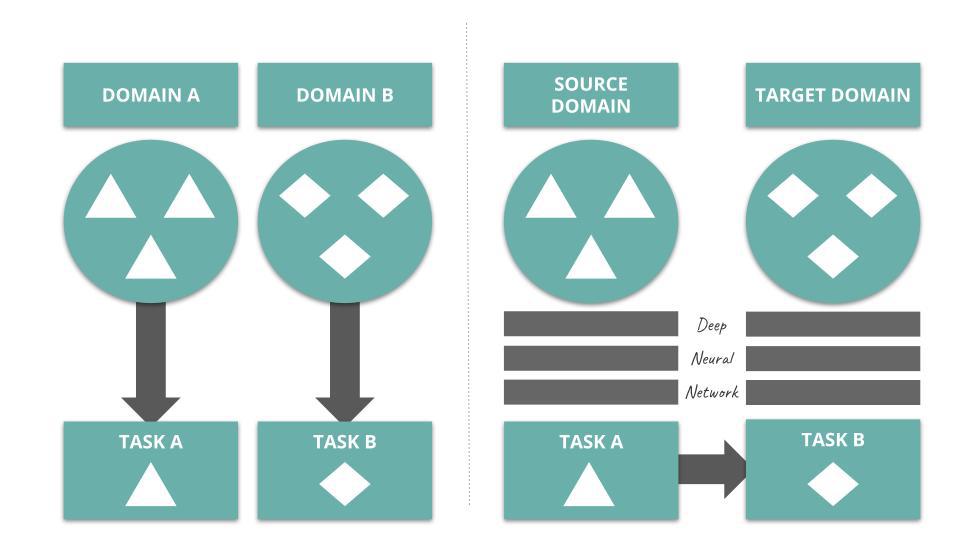
Several studies onboard deep transfer learning into their vHTS design. Check out Kantify’s white paper to learn more about transfer learning in the context of small molecule screening.
Benefits of transfer learning
In practice, data sets available to train models for in silico, or computer-based, drug discovery efforts are often small. Indeed, the sparse availability of labeled data is a major barrier to artificial intelligence-assisted drug discovery. One solution to this problem is to develop algorithms that can cope with relatively heterogeneous and scarce data. Transfer learning can leverage existing, generalizable knowledge from chemical data to enable learning of a separate task from a small set of data. Transfer learning has been used to address low data tasks in many fields such as computer vision, natural language processing, and now drug discovery too. In SMILES Transformer: Pre-trained Molecular Fingerprint for Low Data Drug Discovery, the research team relies on language models to pre-train algorithms on small-molecule data known as SMILES (a list of characters depicting a 2D representation of a molecule) and transfer the knowledge learned to vHTS tasks.
Challenges in transfer learning
The application of transfer learning in drug discovery is still at a preliminary stage, and much remains to be investigated with respect to existing studies. For example, there is no uniform metric to evaluate the performance of transfer learning methods. Thus, it is challenging to make comparisons between applications.
Active learning
What is active learning?
Several strategies exist when it comes to training machine and deep learning algorithms. For instance, active learning relies on iteratively training an algorithm while identifying the most promising next data points. The intuition behind such a tactic is powerful: by combining real-world validation with the predictive force of an AI algorithm, one can find promising drug candidates more quickly with fewer screened compounds.
Because HTS campaigns are costly and take time, and the likelihood of finding a good drug candidate during a campaign can often be low, they are exceedingly well suited to be optimized by a technique like active learning. Rather than being pure in silico process, active learning needs in vitro results to enable iterative training sessions. Reker, 2019 (Practical considerations for active machine learning in drug discovery), depicts the process as follow:
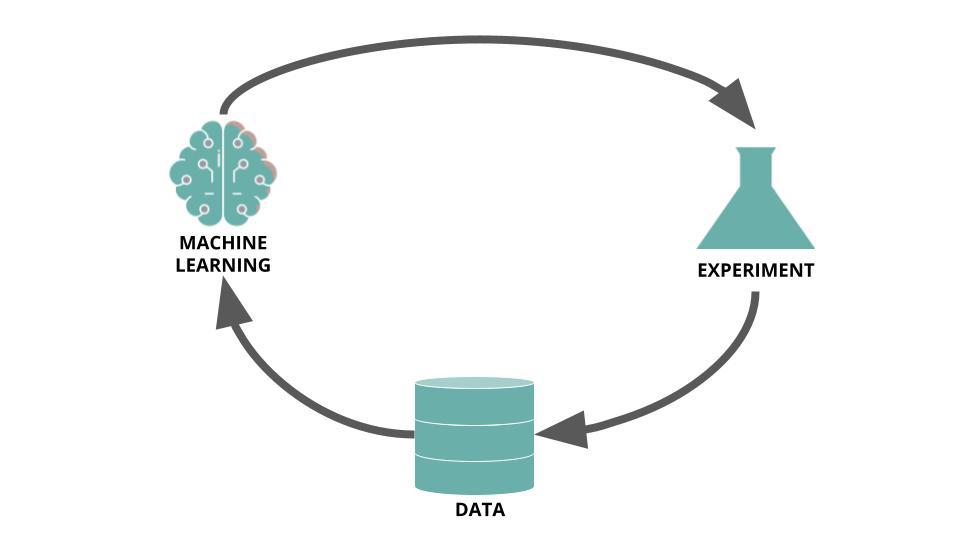
The following insights are largely inspired from Practical considerations for active machine learning in drug discovery) (Reker, 2019).
Benefits of active learning
Active learning relies on finding drug candidates while screening fewer molecules than traditional HTS approaches. Reducing the number of screenings naturally spares time and money in the drug discovery process. Moreover, some bioassays might involve the use of toxic materials, such as nuclear components, or expensive elements such as gold. Thus, active learning is also beneficial when dealing with such resource-heavy experiments.
In his research, Reker admits that only a few studies have compared the ability of active learning pipelines to compete against human optimizations. Yet, the results from Evolving and nano data-enabled machine intelligence for chemical reaction optimization (Reker et al., 2018) and Human versus robots in the discovery and crystallization of gigantic polyoxometalates (Duros et al., 2017) show that active learning campaigns do not only significantly outcompete traditional HTS campaigns, but also lead to more systematic and explorative optimizations. In conclusion, active machine learning in drug discovery should accelerate the drug discovery process and early results show that it can successfully compete against human optimization.
Challenges in active learning
According to Reker, active machine learning has been known for 30 years and tested in drug discovery pipelines for 15 years, but it still has not been widely adopted by companies working on R&D in drugs. Two main barriers stand in the way of larger deployment. First, high throughput screening platforms are designed for large-scale and parallelized testing. Consequently, it might be very difficult to cherry-pick compounds for iterative testing. Second, poor engineering practices within the machine learning part might result in failures.
In How not to develop a quantitative structure-activity or structure-property relationship (QSAR/QSPR), the authors narrow down some of the issues that engineer teams should care about. Here are 4 of them:
- failure to take account of data heterogeneity: having homogenous data might result in good results during testing phases but poor ones in production stages;
- use of inadequate data: small molecule screening is a 3D problem that should not be dealt with 2D descriptors only, for instance;
- overfitting of data: overfitting in machine learning is the process of training an algorithm that does not succeed well in generalizing to new data points;
- inadequate training/test set selection: the notion of chemical scaffolds needs to be taken into account in such a split, for example.
Despite those challenges, Reker believes that active machine learning will become a key technology to guide molecular optimizations in the following years. In particular, other screening technologies than HTS, such as low or modular throughput screening, should work well with active learning: they depart from traditional, rigid, and linear experiment designs like in HTS.
Conclusion
Virtual high throughput screening based on Artificial Intelligence is booming and major breakthroughs are to be expected. In this article, a few key concepts are explored. Deep learning, transfer learning and active learning have been successfully applied in many disciplines and drug discovery processes are starting to be disrupted by those combined technologies.
Kantify is developing technologies that leverage all these techniques in order to accelerate and improve the drug discovery process. You can read up on our work here. For more information, don’t hesitate to contact our CEO, Ségolène Martin (segolene@kantify.com) or our CTO, Nik Subramanian (nik@kantify.com).